Índice
Introducción
Los primeros estudios del síndrome de Klippel-Trenaunay datan de 1900 cuando Klippel y Trenaunay describieron un caso caracterizado por un nevo en el miembro inferior, várices e hipertrofia de los tejidos y del esqueleto del segmento afectado, denominado nevo varicoso osteohipertrófico. Más tarde, en 1918 Parkes Weber describió un caso, similar pero no idéntico, denominado hemangiectasia hipertrófica, que se distinguía por la presencia de angiomas, várices, aneurismas cirsoideos o arteriovenosos e hipertrofia segmentaria.(1)
Concepto
El síndrome de Klippel-Trénaunay, también denominado KTS es una condición que afecta el desarrollo de los vasos sanguíneos, los tejidos blandos y los huesos. Se caracteriza por tres señales: una mancha de nacimiento de color vino de porto, crecimiento excesivo de los tejidos blandos y los huesos y venas varicosas.(2)
El crecimiento excesivo de los huesos y tejidos blandos por lo general comienza en la infancia y muchas veces solamente afecta a una pierna. Sin embargo, también puede afectar los brazos o la parte superior del cuerpo (torso). En la pierna (o brazo) afectada puede haber dolor, sensación de pesadez y dificultad para moverse.(2)
La causa exacta de este síndrome es desconocida. Muchos casos son causados por mutaciones en el gen PIK3CA. En la mayoría de las veces es esporádico, pero en algunos casos es heredado de forma autosómica dominante.(3)
El síndrome de Klippel-Trenaunay (SKT) es una rara enfermedad vascular congénita de causa desconocida, en la cual los vasos sanguíneos y/o linfáticos no se forman correctamente. Se caracteriza por una tríada de síntomas: manchas de vino de oporto (malformación venosa capilar) que cubren uno o más miembros, anomalías vasculares congénitas, generalmente venas varicosas, ausencia o duplicación de una estructura venosa, malformación o hipertrofia (agrandamiento de un miembro). La ausencia de fístula arteriovenosa lo distingue del síndrome de Parkes- Weber.(3)
Los investigadores creen que el síndrome de Klippel-Trénaunay es parte de un grupo de enfermedades relacionadas a mutaciones en el gen PIK3CA que son asociadas con sobrecrecimiento (PROS), que también incluye el síndrome MCAP y el síndrome CLOVE, hemimegalencefalia, hiperplasia fibroadiposa y nevo epidérmico.(2,4)
La mayoría de los casos son esporádicos lo que significa que ocurren en personas sin otros casos en su familia, debido a que es causada por mutaciones somáticas, que no afectan las células germinales (óvulo o espermatozoide). Los casos familiares que se han reportado en la literatura no han sido confirmados. De esta forma, si una persona tiene SKT el riesgo de tener un(a) hijo(a) con la enfermedad es similar al resto de la población.
El tratamiento es sintomático y de apoyo.(2,4)
Muchos casos del SKT pueden ser causados por mutaciones en el gen PIK3CA. Estas mutaciones son somáticas, o sea, ocurren después de la concepción (después de que el óvulo y el espermatozoide se unen), en las células primitivas que forman un miembro que estaban destinados a convertirse en sangre, vasos linfáticos, grasa, y huesos. Debido a que esta mutación no se produce en las células germinales (óvulo o espermatozoide), no se puede transmitir en una familia (es decir, no se heredan). Además, como las mutaciones somáticas solamente ocurren en algunas células del cuerpo, pero no en todas, las señales y síntomas se limitan a las áreas del cuerpo donde las células tienen mutaciones.(1,2)
El gen PIK3CA gen proporciona instrucciones para hacer una proteína que es parte de una enzima importante para el crecimiento y la división celular (proliferación), el movimiento (migración) de las células, y la supervivencia celular. Las mutaciones en el gen PIK3CA originan una enzima anormal que resulta en que las células crezcan y se dividan de forma continua, lo que lleva al crecimiento anormal de los huesos, los tejidos blandos, y los vasos sanguíneos. (1,2)
Algunos investigadores creen que todos los casos del SKT son causados por mutaciones en el gen PIK3CA, y que cuando no se encuentra una mutación en este gen, es porque la persona tiene otra enfermedad diferente.[4] Sin embargo, otros investigadores creen que existen mutaciones en otros genes que todavía no se han identificado que causan el SKT.(1,2)
Síntomas (2,4–6)
La mayoría de los pacientes demuestran los 3 signos de este síndrome:
- Hemangioma o nevus flammeus (mancha de color de vino de porto): Presente en 98% de los casos, localizado unilateralmente (frecuentemente en las piernas, nalgas, abdomen y región inferior del tronco) sin envolver la línea media del cuerpo. Tiene profundidad variable pudiendo afectar únicamente a la piel o envolver el tejido debajo de la piel (tejido subcutáneo), músculos y huesos. Los órganos internos, como la pleura, bazo, el hígado, la vejiga y el colon también pueden ser afectados, lo que puede llevar a sangramientos internos.
- Venas varicosas: Presentes al nacer (congénitas) pero que pueden no ser notadas hasta que el niño comienza a caminar. Las varices pueden ser extensas y normalmente se sitúan debajo de la rodilla, o lateralmente por encima de la rodilla y en otros casos en la región pélvica. Las varices pueden afectar los sistemas venosos superficiales y profundos o diversos órganos. Pueden permanecer estables o aumentar con el tiempo llevando a dolor y linfedema (que empeoran durante el embarazo en las mujeres afectadas).
- Hipertrofia (aumento de tamaño): Del tejido óseo y tejidos blandos en un miembro que puede ser notada al nacer y que progresa durante los primeros años de vida. La hipertrofia del miembro puede ser secundaria a aumento en la largura del miembro (crecimiento del hueso) o un aumento de la circunferencia (crecimiento de los tejidos blandos).
Puede haber también malformaciones linfáticas entre las que se incluyen:
- Obstrucción linfática
- Linfedema
- Malformaciones linfáticas profundas que pueden llevar a problemas en órganos internos y desfiguramiento de los genitales.
- Otras señales y síntomas que han sido descritos son:
- Asimetría del rostro
- Anomalías en los dientes
- Cabeza muy grande (macrocefalia) o muy pequeña (microcefalia)
- Anomalías de los dedos como dedos extra (polidactilia), dedos pegados (sindactilia) o falta de dedos (oligodactilia)
- Sensaciones de quemadura o pinchazos (parestesias)
- Descalcificación de los huesos afectados
- Insuficiencia venosa crónica
- Dermatitis por estasis
- Mala cicatrización de heridas
- Úlceras
- Trombosis
- Embolia
- Dolor en los huesos.
- En casos más raros se ha descrito como Discapacidad intelectual y/o convulsiones (generalmente cuando hay un hemangioma en la cara).
Con menor frecuencia puede encontrarse claudicación intermitente, úlceras varicosas, aumento de la temperatura cutánea, caída del cabello difusa, disqueratosis, alteraciones en la sudoración, producción de lágrimas y saliva. También se han observado alteraciones de la marcha y asociación con hemihipertrofa de la cara y linfangioma cutáneo, venas varicosas pulmonares y complicaciones orales.(6)
Actualmente muchos investigadores piensan que cuando hay malformaciones en las arterias asociadas, además de las malformaciones venosas (arteriovenosas) los pacientes tienen otra enfermedad que se llama síndrome de Parkes Weber.
Diagnóstico (2,4–6)
El SKT se caracteriza por la presencia de malformaciones capilares (mancha de nacimiento de color vino de porto), crecimiento excesivo de los tejidos blandos y los huesos (hipertrofia) y venas varicosas.
El diagnóstico del síndrome de Klippel-Trénaunay comienza con un examen físico. La derivación a un especialista en malformaciones vasculares es útil para las recomendaciones de evaluación y tratamiento. Durante la evaluación, es preciso la realización de preguntas sobre tu historia clínica y antecedentes familiares, llevar a cabo un examen para detectar hinchazón, venas varicosas y manchas de vino de oporto en la piel y la evaluación visual del crecimiento de los huesos y tejidos blandos.
Mediante diversas pruebas de diagnóstico, es posible evaluar e identificar el tipo y la gravedad de la enfermedad, y por lo tanto determinar el tratamiento. Algunas pruebas son las siguientes:
- Exploración doble. Esta prueba utiliza ondas sonoras de alta frecuencia para crear imágenes detalladas de los vasos sanguíneos.
- Escanografía o fotografía de escáner, esta técnica radiográfica ayuda a ver imágenes de huesos y medir su longitud.
- Resonancia magnética y angiografía por resonancia magnética. Mediante estos procedimientos es posible diferenciar entre hueso, grasa, músculo y vasos sanguíneos.
- Exploración por tomografía computarizada. Una tomografía computarizada crea imágenes tridimensionales del cuerpo que ayudan a detectar coágulos de sangre en las venas.
- Flebografía de contraste. Este procedimiento consiste en inyectar un tinte en las venas y tomar radiografías que puedan revelar venas anormales, obstrucciones o coágulos de sangre.
Los criterios diagnósticos fueron revisados por un grupo de investigadores que dividen los síntomas hallados en los afectados por el SKT en 2 grupos (Grupo A y Grupo B). Para una persona ser diagnosticada con el SKT debe tener dos hallados mayores (por lo menos un hallado del grupo A, que siempre debe incluir el hallado 1 o 2, y por lo menos un hallado del grupo B):
Grupo A - Malformaciones vasculares presentes al nacer
Hallado 1: Malformaciones capilares, que incluye la mancha de color vino de porto.
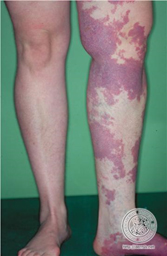
Hallado 2: Malformaciones venosas que pueden ser: venas muy pequeñas o ausentes, persistencia de venas fetales, varices, aumento de tamaño de las venas, venas tortuosas y malformaciones valvulares de las venas.
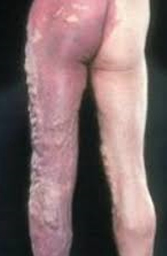
Hallado 3: Malformaciones arteriovenosas. Incluye solamente las malformaciones arteriovenosas muy pequeñas o fistulas arteriovenosas. Se cree que cuando hay malformaciones arteriovenosas en realidad se trata de otra enfermedad llamada síndrome de Parkes Weber.
Hallado 4: Malformaciones linfáticas. Localización: las manchas de color vinieron de porto pueden estar situadas en cualquier lugar del cuerpo, pero son raras en la cara; las malformaciones venosas o linfáticas están localizadas principalmente en las extremidades cerca del tronco (muslo) en la pelvis o en el hombro, pero pueden estar en cualquier lugar menos en la cara o en el cerebro.
Grupo B – Crecimiento anormal
Hallado 1: Crecimiento anormal en la longitud o grosor del hueso.
Hallado 2: Crecimiento anormal del tejido blando en la longitud o grosor.
El crecimiento anormal incluye:
- Tamaño aumentado (hipertrofia): Frecuentemente de una pequeña parte del cuerpo (como el aumento del tamaño del dedo o “macrodáctilia”) o de parte mayor del cuerpo (una extremidad entera o la mitad del cuerpo).
- Tamaño disminuido (hipotrofia): de una parte, pequeña o mayor del cuerpo. No es tan común.
- Localización: La alteración del crecimiento puede estar presente tanto en el mismo sitio que la malformación vascular (más comúnmente) o en otro sitio (menos comúnmente).
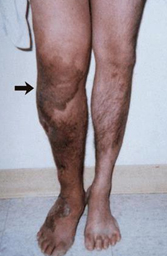
Más o menos 63% de los pacientes diagnosticados presentan los tres síntomas. El síntoma más común, presente en 98% de los pacientes, es la mancha color de vino de porto. En muchos casos esta mancha está situada en el miembro (especialmente el miembro inferior) que es mayor. Cuando la mancha está situada en el tronco rara vez cruza la línea media del cuerpo. Las venas varicosas también están presentes en la mayoría de los pacientes con síndrome de Klippel-Trénaunay, y se notan más durante la adolescencia cuando, en algunos casos, pueden aumentar, causando dolor, linfedema, tromboflebitis y úlceras. La hipertrofia está presente al nacimiento y normalmente resulta de ectasia venosa y afecta los huesos, los tejidos blandos, o ambos. Casi siempre la hipertrofia es de un miembro inferior (muslo o pierna y pie).
En muchos casos, una historia completa y examen físico son todo lo que se requiere para diagnosticar el SKT. Sin embargo, en otros casos, y cuando hay complicaciones, los estudios de imagen pueden ser útiles, sobre todo la realización de angiografía, para buscar fistulas arterio-venosas y diferenciar entre el síndrome de Klippel Trenaunay y el síndrome de Parker Weber.
Los diagnósticos diferenciales incluyen aquellas malformaciones que generan sobrecrecimiento y malformaciones vasculares como los síndromes de Proteus, de Sturge-Weber, de Parkes-Weber, de Maffuci, CLOVES, la anomalía fibroadiposovascular (FAVA) y Síndrome Tumor hamartoma PTEN.
Tratamiento (6,8)
El tratamiento es dirigido a las señales y síntomas que se presenten. La compresión elástica es indicada para los casos de insuficiencia venosa crónica, linfedema, celulitis y sangramientos. En algunos casos se puede hacer cirugía con láser o cirugías ortopédicas más extensas.
Entre los tratamientos pueden incluirse los siguientes:
- Terapia de compresión. Las extremidades afectadas se envuelven con vendas o tela elástica para ayudar a evitar la inflamación, los problemas de venas varicosas y las úlceras en la piel. Estas vendas o tela elástica por lo general deben ser hechas a medida. Pueden usarse dispositivos de compresión neumática intermitente (mangas para los brazos o las piernas que se inflan y desinflan automáticamente a intervalos establecidos).
- Los masajes, la compresión y el movimiento de las extremidades, según corresponda, pueden ayudar a aliviar el linfoedema en los brazos o las piernas, y la inflamación de los vasos sanguíneos.
- Dispositivos ortopédicos. Entre ellos se puede incluir calzado ortopédico o plantillas para compensar la diferencia en el largo de la pierna.
- Este es un procedimiento quirúrgico ortopédico que puede detener de manera eficaz el crecimiento excesivo del largo de la extremidad inferior.
- Embolización. Este procedimiento, que se realiza mediante catéteres pequeños que se colocan en las venas o las arterias, bloquea el flujo de sangre a ciertos vasos sanguíneos.
- Terapia con láser. Este procedimiento puede usarse para aclarar las manchas en vino de oporto en la piel y para tratar ampollas tempranas en la piel.
- Ablación de las venas por radiofrecuencia o láser. Este procedimiento mínimamente invasivo se usa para cerrar las venas anormales.
- Se inyecta en una vena una solución que crea tejido cicatricial que ayuda a cerrar la vena.
- Cirugía. En algunos casos, la reconstrucción o extracción quirúrgica de las venas afectadas, la extracción del tejido excedente y la corrección del crecimiento excesivo del hueso pueden ser beneficiosos.
- Investigaciones tempranas indican que un medicamento llamado sirolimus (Rapamune) puede ayudar a tratar las malformaciones vasculares complejas sintomáticas, pero puede que provoque efectos secundarios significativos y se necesitan más estudios.
La cirugía de láser puede disminuir o erradicar algunas lesiones en la piel.
La cirugía ortopédica se hace para corregir diferencias en el tamaño de los miembros, sin embargo, en los casos más leves no es necesaria y el problema es resuelto con el uso de aparatos ortopédicos.
Es importante destacar la morbilidad y mortalidad de esta enfermedad provocada por el tromboembolismo desencadenado por la estasis venosa, el cual puede evitarse cerrando oportunamente las venas embriológicas. Las venas marginales pueden ser obliteradas mediante embolización, fotocoagulación con láser endovenoso o se las puede remover quirúrgicamente (se la recomienda tempranamente en la infancia para evitar que alcancen un gran tamaño y por las tortuosidades). Las malformaciones linfáticas de tamaño importante provocan discapacidad, pudiendo realizarse cirugía de citoreducción. Las deformidades óseas requieren de seguimiento ortopédico; el sobrecrecimiento óseo de extremidades puede ser abordado con epificiodesis cuando ocasionan gran discrepancia. Las malformaciones capilares pueden ser tratadas con láser, las vesículas linfáticas cutáneas con escleroterapia, fotoevaporación láser con dióxido de carbono, cauterización o escisión. La evaluación hematológica es necesaria para descartar trombofilias. Las complicaciones infecciosas recibirán tratamiento oportuno con antibióticos. Estos pacientes además deben recibir acompañamiento psicológico
Como equipo de enfermería especializado en curación de úlceras, vamos a centrarnos en los cambios a nivel vascular provocados por el síndrome Klippel- Trenaunay; (6) KTS debe distinguirse del síndrome de Parkes Weber, diferencia que se define por la ausencia de fístula arteriovenosa lo distingue del síndrome de Parkes- Weber, es decir si hubiese una fistula arteriovenosa sería diagnosticado como el síndrome de Parkes- Weber; una malformación vascular combinada de flujo rápido con sobrecrecimiento de las extremidades. El tratamiento individualizado es necesario para KTS y debe centrarse en el tratamiento de los síntomas.
Por su parte, este síndrome de Parkes-Webes se identifica por una o más malformaciones arteriovenosas de alto flujo, que conlleva a un crecimiento excesivo del hueso y los tejidos blandos de la extremidad afectada, generalmente más grave que en el SKT, y múltiples fístulas arteriovenosas (conexiones anormales entre las arterias y las venas) que pueden conducir a insuficiencia cardíaca. Suele ocasionar dolor en la extremidad afectada y una diferencia de tamaño entre los miembros; asimismo, afecta a los miembros superiores más comúnmente que SKT, pero también a los inferiores.(1,8)
Su ocurrencia es más rara que SKT, pero ambos se incluyen entre los síndromes neurocutáneos con afección vascular.
En el síndrome de Klippel-Trenaunay (KTS), el manejo de una herida en la extremidad afectada puede ser difícil debido a las malformaciones vasculares subyacentes presentes, así como precisa de un tratamiento adicional significativo y la extensión de la estancia hospitalaria, por lo tanto deben anticiparse complicaciones de la herida en pacientes con síndrome de Klippel-Trenaunay sometidos a procedimientos quirúrgicos ortopédicos, especialmente amputaciones terminales.(9)
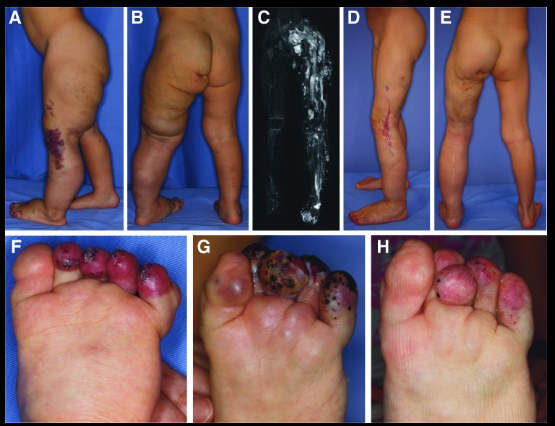
En general, las venas varicosas en pacientes con KTS se manejan de manera conservadora con descanso intermitente, elevación de extremidades y recordatorios continuos de la importancia de usar prendas de compresión graduadas. La tromboflebitis en las venas varicosas debe tratarse con analgésicos y antiinflamatorios.
El tratamiento inicial del linfedema comienza con la educación del paciente y la familia sobre la higiene de la piel, con el objetivo final de minimizar las infecciones de la piel.
El tratamiento inapropiado, como la eliminación de venas varicosas funcionales o la reducción quirúrgica, puede dañar la circulación venosa y linfática ya deteriorada y empeorar aún más el linfedema, por lo que el diagnóstico temprano y el tratamiento adecuado con prendas de compresión graduada pueden prevenir o retrasar el curso del linfedema en pacientes con KTS. Estas prendas también pueden proteger la extremidad de traumas menores.
Se han informado varias opciones para el tratamiento local de las úlceras cutáneas incluida la terapia de heridas con presión negativa, terapia con láser de bajo nivel, y la aplicación tópica de factor de crecimiento recombinante derivado de plaquetas humano BB.
Bibliografía
- Ortiz Limonta D, Gómez Jurado A, Sánchez Figueredo SA. Síndrome de Parkes Weber en una infante. MEDISAN. octubre de 2016;20(10):2276-81.
- Síndrome de Klippel-Trenaunay-Weber: antecedentes, fisiopatología, etiología [Internet]. [citado 23 de junio de 2020]. Disponible en: https://reference.medscape.com/article/1084257-overview
- The Pathology of Vessels – Phat N. Vuong, Colin Berry – Google Libros [Internet]. [citado 24 de junio de 2020]. Disponible en: https://books.google.es/books?id=XNwFCAAAQBAJ&pg=PA386&lpg=PA386&dq=Klippel+M,+Tr%C3%A9naunay+P.+Du+nevus+variqueux+ost%C3%A9o+hypertrophique.+Arch+Gen+Med+1900;+185:+641-72.&source=bl&ots=EBmO_z5To5&sig=ACfU3U0w8LUKMcz4VST7VcVqhQJ54Z_dag&hl=es&sa=X&ved=2ahUKEwi-iaSCiZvqAhUCDWMBHUxFA6QQ6AEwAHoECAoQAQ#v=onepage&q=Klippel%20M%2C%20Tr%C3%A9naunay%20P.%20Du%20nevus%20variqueux%20ost%C3%A9o%20hypertrophique.%20Arch%20Gen%20Med%201900%3B%20185%3A%20641-72.&f=false
- Oduber CEU, van der Horst CMAM, Hennekam RCM. Klippel-Trenaunay syndrome: diagnostic criteria and hypothesis on etiology. Ann Plast Surg. febrero de 2008;60(2):217-23.
- Síndrome de Klippel-Trénaunay | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program [Internet]. [citado 23 de junio de 2020]. Disponible en: https://rarediseases.info.nih.gov/espanol/12255/sindrome-de-klippel-trenaunay
- Gontero R, Ortiz A, Roverano S, Paira S. Síndrome Klippel-Trenaunay: comunicación de dos casos. :6.
- Síndrome de Klippel-Trénaunay – Síntomas y causas – Mayo Clinic [Internet]. [citado 24 de junio de 2020]. Disponible en: https://www.mayoclinic.org/es-es/diseases-conditions/klippel-trenaunay/symptoms-causes/syc-20374152
- Ishikawa K, Yamamoto Y, Funayama E, Furukawa H, Sasaki S. Wound-Healing Problems Associated with Combined Vascular Malformations in Klippel-Trenaunay Syndrome. Adv Wound Care. 1 de junio de 2019;8(6):246-55.
- Gates PE, Drvaric DM, Kruger L. Wound healing in orthopaedic procedures for Klippel-Trenaunay syndrome. J Pediatr Orthop. diciembre de 1996;16(6):723-6.

